Регуляция экспрессии генов на уровне транскрипции
Регуляция экспрессии генов на уровне транскрипции осуществляется с помощью сигнальных последовательностей молекул ДНК и белковых комплексов, в частности транскрипционных факторов. Для разных генов системы контроля экспрессии выглядят по-разному, - с этой точки зрения существуют индуцибельные гены, экспрессирующиеся в ответ на определённые стимулы и преимущественно в определённых типах клеток, и гены «домашнего хозяйства», экспрессирующиеся практически постоянно и в разных типах тканей (например, гены рибосомальных белков; ферментов, участвующих в окислительно-восстановительных процессах и т.п.). Для первых контроль экспрессии выглядит более сложно.
Транскрипция генов эукариот осуществляется тремя РНК-полимеразами, причём промоторы для каждой из них характеризуются различными регуляторными последовательностями нуклеотидов, с которыми взаимодействуют различные факторы транскрипции, влияющие на уровень транскрипции соответствующих генов [7]. Факторы транскрипции у эукариот обладают консервативными доменами, обеспечивающими высокоспецифичные взаимодействия видов «белок – белок» и «белок – нуклеиновая кислота» [8]. При этом может происходить как активация транскрипции (в таком случае факторы называют активаторами, а осуществляемую ими регуляцию - позитивной), так и блокада считывания РНК-полимеразами генетической информации, например, путём создания стерических препятствий для взаимодействия активаторов транскрипции и регуляторных последовательностей ДНК (негативная регуляция). Факторы транскрипции в клетке объединяются вместе с другими регуляторными белками в регуляторные комплексы. Возможны разные сочетания белковых факторов, что придаёт каждому из образующихся функциональных комплексов уникальные свойства путём изменения специфичности взаимодействия комплекса с регуляторными последовательностями ДНК [9].
|
|
|
Эукариоты способны использовать для регуляции транскрипции изменение структуры хроматина. Это играет существенную роль в процессах дифференцировки клеток и поддержании их функционирования. При этом могут осуществляться как репрессия, так и дерепрессия отдельных генов, их массивов и даже целых хромосом [10], например, дезактивация X-хромосомы и образование тельца Барра.
Посттранскрипционная регуляция экспрессии генов
Посттранскрипционная регуляция осуществляется на уровне мРНК в период после окончания их синтеза и до начала трансляции. Этот этап можно разделить на несколько подэтапов или разновидностей:
- модификации предшественников зрелой мРНК (альтернативный сплайсинг, кэпирование, присоедиение поли-А-конца);
|
|
|
- транспорт и депонирование зрелой мРНК;
- РНК-сайленсинг;
- деградация мРНК.
Эти процессы переходят друг в друга, между ними нет чёткой границы, и кроме того, они зачастую скоординированы и с вышестоящими механизмами контроля над экспрессией. К примеру, у дрожжей наблюдается определённая корреляция процессов транскрипции, модификации гистонов и РНК-интерференции, в результате чего происходит инактивация генной экспрессии [11]. Инициация сайленсинга с помощью метилирования ДНК зависит от соотношения микро-РНК и её мишени [12].
Актуально на данный момент изучение РНК-интерференции, отвечающей за процессы сайленсинга, деградации мРНК. МикроРНК выступают в качестве регуляторов генной экспрессии на разных этапах функционирования клетки, ткани, органа. Каждая микроРНК может регулировать экспрессию множества различных мРНК [13], [14], [15]. Механизм регуляции экспрессии при помощи микроРНК до конца ещё не выяснен: происходит ли репрессия на уровне трансляции или же мРНК подвергается деградации ещё до начала этого процесса. Вероятно, у животных (в отличие от растений) репрессия с помощью микроРНК может осуществляться несколькими способами и на разных уровнях – инициации, элонгации, с помощью преждевременной терминации трансляции и деградации образующихся пептидов [16], [17], [18], [19], [20].
|
|
|
МикроРНК играет значительную роль в процессах развития, дифференцировки и функционирования нервной ткани организма [21].
1.1.2. Проблемы изучения транскриптома
Транскриптом – это совокупность продуктов транскрипции, т.е. молекул РНК, синтезированных клеткой или группой клеток в определённый период их функционирования. Раздел молекулярной биологии, изучающий транскриптом, называется транскриптомикой. Транскриптомика изучает структуру транскриптов, их пространственное и временное разделение в клетке, определяет уровень их экспрессии. Уровень мРНК гена определяется комбинацией процессов инициации и элонгации транскрипции, процессинга и деградации мРНК, в связи с чем возникает вопрос, какой вклад вносит каждый из этих процессов, как это связано с функцией соответствующего белка, и главное – как уровень мРНК гена коррелирует с уровнем соответствующего белка.
Экспрессия генов обусловлена большим количеством факторов, и эта многофакторность приводит к колебаниям уровня мРНК (и в ряде случаев белка) между клетками одной популяции (изогенными клетками) – транскрипционному шуму [22]. К этим факторам относится в том числе взаимодействие между клетками. Такую изменчивость следует отличать от изменчивости в ответ на целенаправленный стимул (пластичности экспрессии) [23]. Транскрипция генов происходит всплесками – короткий период транскрипционной активности сменяется периодом инактивации. Промежутки между всплесками нерегулярны. Причины этого до конца не ясны: есть предположения, что они кроются в ремоделировании хроматина, приводящем к транскрипционной активности гена [22], [24]; в формировании преинициаторных комплексов в области промотора ДНК, обеспечивающих работу РНК-полимеразы II – они существуют в течение короткого времени, что приводит к пульсирующей транскрипции [25]. Возможны другие причины вариабельности уровня мРНК – например, неравномерное разделение клеточного содержимого в результате митоза [26].
|
|
|
Все эти факты нужно обязательно учитывать при количественных исследованиях мРНК. Разные гены, кроме того, могут характеризоваться разным уровнем транскрипционного шума.
В связи с этим возникает вопрос, как соотносится уровень мРНК гена и уровень кодируемого этим геном белка в клетке. Ситуацию усложняет тот факт, что короткоживущие мРНК могут приводить к синтезу долгоживущих белков и наоборот.
Ряд авторов утверждает, что корреляция как таковая между уровнем белка и соответствующей ему мРНК отсутствует [27], [28]. Однако по результатам исследования Schwanhausser с соавторами, основанном на методе параллельной импульсной маркировки белков радиоактивно-меченными аминокислотами и РНК, меченной 4-тиоуридином (метод позволяет одновременно определять внутриклеточный оборот белков и мРНК), определённая корреляция была найдена. В соответствии с этим гены были разделены на 4 группы:
- стабильные мРНК, стабильные белки (гены ферментов гликолиза, глюконеогенеза, клеточного дыхания);
- нестабильные мРНК, нестабильные белки (гены, продукты которых участвуют в регуляции клеточного цикла, спирализации и деспирализации хроматина, факторы транскрипции);
- нестабильные мРНК, стабильные белки (продукты данных генов участвуют, в частности, в процессинге РНК);
- стабильные мРНК, нестабильные белки (продукты данных генов – секреторные белки; ферменты, участвующие в поддержании гомеостаза, многие гидролазы, белки иммунного ответа) [29].
Иначе говоря, существует определённая (частичная) корреляция уровня белка и уровня соответствующей мРНК, но эта корреляция зависит от функции белка, и она, вероятно, тем выше, чем выше уровень транскрипционной активности гена [30].
Определение уровня мРНК и соответствующих белков – необходимая часть исследований в функциональной геномике, поскольку недостаточно знаний о структуре гена, его транскрипта или транскриптов и специализации соответствующего белка для того, чтобы можно было судить об участии (степени участия) гена в физиологических процессах или в развитии патологий.
1.2. Церебральная ишемия
Церебральная ишемия (ишемический инсульт) – это острое нарушение мозгового кровообращения с последующим повреждением ткани мозга и нарушением его функций, а затем и гибелью участка мозга (мозговым инфарктом). Причинами могут быть тромбоз или эмболия (закупорка артерий), приводящие к уменьшению поступления кислорода и глюкозы из кровеносного русла к клеткам нервной ткани, что нарушает протекание окислительно-восстановительных процессов (и как следствие - всех остальных процессов) в них. На это сразу же реагируют клеточные структуры. В свою очередь на изменение внутриклеточных процессов реагирует клеточное ядро, и начинается экспрессия генов, обеспечивающих синтез веществ, участвующих в процессах как разрушения, так и восстановления.
1.2.1. Реакция тканей на снижение уровня кровотока
Оптимальным уровнем кровотока является 50 - 60 мл на 100 г мозгового вещества в минуту. При более низкой скорости наблюдаются следующие нарушения обменных процессов в нервной ткани:
- при кровотоке ниже 50 - 55 мл на 100 г/ мин. в условиях снижения скорости окислительно-восстановительных процессов (вследствие недостатка кислорода и глюкозы, поступающих с кровотоком) происходит резкое падение уровня аденозинтрифосфата (АТФ). Вследствие этого, в частности, происходит общее торможение синтеза белков в нейронах. Наблюдается избирательная экспрессия определённых генов [31];
- при снижении кровотока до 35 мл на 100 г/мин. и ниже в условиях недостатка кислорода происходит активация анаэробного гликолиза (образуется всего 2 молекулы АТФ за один оборот фермента), повышение концентрации молочной кислоты (лактат-ацидоз), цитотоксический отёк;
- в условиях снижения уровня АТФ отмечается дисфункция ионных каналов активного транспорта, деполяризация мембран, выброс возбуждающих нейротрансмиттеров, в частности глутаминовой кислоты;
- при прогрессирующем снижении кровотока наблюдается недостаточность нейрональной электрической активности [32];
- в условиях того же дефицита АТФ (вследствие замедления окислительно-восстановительных процессов) проявляется прогрессирующая недостаточность энергетически зависимых ионных насосов, которые сохраняют относительное постоянство внутриклеточной среды;
- в условиях снижения кровотока до 10 - 15 мл на 100 г вещества в минуту начинается неконтролируемое проникновение ионов через клеточную мембрану нейрона; идёт каскад разрушительных внутриклеточных процессов. Через некоторое время происходит гибель нейронов [33]. До запуска этой стадии нейроны остаются жизнеспособными и могут восстановить свои функции в случае восстановления кровоснабжения.
В первые несколько часов ядро ишемии окружено ещё живой тканью (так называемая "ишемической полутень", или "пенумбра"), в которой наблюдаются функциональные изменения, но структурные изменения пока отсутствуют [34], [35]. Кровоснабжение здесь ниже уровня, необходимого для нормального функционирования, но выше критического порога необратимых изменений. Гибель клеток в зоне "полутени" приводит к расширению области инфаркта. Именно эта зона является основной "мишенью" при лечении ишемического инсульта в первые часы заболевания (предпочтительно первые 3 - 6 часов). Длительность существования зоны ишемической полутени индивидуальна и зависит от метаболизма мозга и состояния нейроэндокринной и иммунной систем, в среднем это около 50 часов [36], [37], [38], [39], [40], [41], [42], [43].
Процессы при ишемии сопровождаются развитием отёка мозга. Он развивается через несколько минут после развития локальной ишемии. Причина - нарушение функционирования мембранных белков, неконтролируемое проникновение ионов через мембраны клеток, вслед за которыми проникает и вода. В свою очередь данное состояние возникает вследствие недостатка АТФ и соответственно энергетических возможностей для поддержания гомеостаза. Сначала развивается цитотоксический (внутриклеточный) отёк, а затем, по причине гибели клеток гематоэнцефалического барьера, и внеклеточный, сопровождаемый накоплением в зоне повреждения продуктов анаэробного гликолиза [44], [45], [46], [47]. С повреждением гематоэнцефалического барьера в ткани мозга проникают лейкоциты, вызывающие в том числе гибель здоровых клеток нервной ткани [48], [49]. При этом увеличивается объём мозга, повышается внутричерепное давление. Это может повлечь за собой смещение частей головного мозга друг относительно друга. Смерть больных часто происходит по причине сдавления нижних отделов продолговатого мозга [50].
1.2.2. Клеточные реакции при ишемии головного мозга
При развитии острой фокальной ишемии происходит запуск патологических биохимических процессов во всех типах клеток нервной ткани, вызывающих нейрональные нарушения, астроцитоз (повышенное содержание астроцитов), микроглиальную активацию, а также изменения нейтрофилов, макрофагов, эндотелиальных клеток [51].
В области ишемической полутени (пенумбры) раньше и в большей степени поражаются глиальные клетки, чем нейроны коры головного мозга [52]. Глиальные клетки количественно значительно преобладают над нервными, занимая объём между сосудами и нейронами, причём нейроны настолько тесно окружены нейроглией, что иногда нелегко отделить одну фракцию от другой. Такая тесная взаимосвязь нейронов и глии является основой для их физиологических взаимодействий. Глия выполняет не только трофическую и защитную функции, но и способна тормозить гиперактивность нейронов, а также участвовать в регуляции энергетического потока при активации нейронов, окисляя глюкозу с выделением лактата. Астроциты способны захватывать и метаболизировать глутаминовую кислоту - медиатор возбуждения, способный при высокой концентрации оказывать цитотоксическое действие на нейроны. Глия участвует в синтезе цитокинов (пептидных молекул, регулирущих межклеточные взаимодействия, стимуляцию и подавление роста клеток, дифференциацию, функциональность, ответ на повреждающие воздействия и апоптоз), цГМФ (циклогуанозинмонофосфата, выступающего в качестве посредника передачи сигнала, в регуляции клеточного цикла), NO (оксида азота (II), обладающего разнообразным биологическим действием). Также глия принимает участие в синтезе глиальных факторов роста нейронов, участвующих в репарации нейронов и их трофике [53].
По результатам исследований на моделях экспериментального ишемического инсульта у крыс была установлена последовательность реакций разных типов клеток мозга на ишемию. Существуют различия в развитии процессов в мозге грызунов и приматов, однако многочисленные общие черты позволяют частично экстраполировать полученные данные на понимание механизма развития ишемии у человека. В первую очередь при ишемии мозга наблюдается изменение глиальных клеток. В случае астроцитов наблюдается их набухание, фрагментация отростков, дезинтеграция. При этом снижается экспрессия астроцитарного маркера - кислого глиального фибриллярного белка GFAP [52], [54]. Однако через 4 - 6 часов наблюдается активация астроцитов и повышение экспрессии GFAP. Примерно через 24 часа вокруг ишемического ядра образуется сеть GFAP-позитивных астроцитов. В конечном итоге это ведёт к образованию глиального рубца через 5 - 10 дней после начала ишемии [55].
Наблюдаются и изменения микроглиальных клеток. Микроглия - представительство иммунокомпетентных клеток, защищающих мозг от вредных факторов [56], [57]. В нормальных условиях микроглиальные клетки имеют ветвистую отростчатую форму [58] и находятся в состоянии функционального покоя. Они способны на короткое время активироваться, выделяя токсичные провоспалительные медиаторы, но быстро возвращаются в состояние покоя [59]. При ишемии и некоторых других патологических состояниях они втягивают отростки, принимая амёбовидную форму, при этом активируясь и не возвращаясь в состояние покоя, а продолжая синтезировать токсичные для клеток нервной ткани соединения. При этом поддерживается воспалительная реакция, происходят гибель нейронов и ухудшение микроциркуляции, повреждается гематоэнцефалический барьер [60], [61]. Особенно сильно эти нарушения выражены в области пенумбры [62], [63]. С течением времени область локализации активированной микроглии расширяется, и к 5-му дню изменённые клетки микроглии наблюдаются в областях, отдалённых от очага ишемии [63], [64], [65].
Первые изменения в нейронах - их сморщивание - наблюдаются через 30 минут после проведения операции по перевязке артерии. Ещё через час наблюдается увеличение количества гетерохроматина (что говорит о затормаживании экспрессии ряда генов), расширение ЭПР, вакуолизация, набухание митохондрий. Эти изменения сохраняются на протяжение примерно 6 часов и являются обратимыми. Через 10 - 12 часов в ядре ишемии обнаруживаются признаки необратимого повреждения нейронов: разрушение мембранных структур, отложение солей кальция на внутренней мембране митохондрий [52], [66], в зоне ишемической полутени изменения сохраняются дольше. Первые погибшие нейроны обнаруживаются в тканях мозга в среднем через 50 часов после инсульта.
Наряду с клетками, подвергшимися некротической гибели, вдоль внутренней границы зоны ишемического ядра обнаруживаются клетки, подвергающиеся запрограммированной клеточной гибели – апоптозу [67], [68], [69], [70], [71]. Максимальное количество таких клеток появляется на вторые сутки после инсульта, затем их количество снижается [72], но тем не менее апоптозные нейроны продолжают появляться в зоне полутени и спустя 4 недели после начала заболевания.
Через 6-8 часов после начала заболевания наблюдаются изменения нейтрофилов в микроциркуляторном русле. Это связано с активацией микроглии и повышением синтеза противовоспалительных факторов и молекул клеточной адгезии: иммуноглобулин-подобных, интегринов, селектинов и т.п. Нейтрофилы прилипают к эндотелию капилляров, проникают через гематоэнцефалический барьер и внедряются в ишемизированную ткань. Максимум инфильтрации нейтрофилов в ткани мозга наблюдается через 48 - 96 часов с момента наступления инсульта, после чего их количество постепенно снижается [73], [74], [75]. Помимо нейтрофилов в ишемизированную ткань проникают макрофаги, но максимум инфильтрации макрофагами наступает несколько позже - на 5-е - 7-е сутки после начала инсульта [76]. С этого момента ишемизированная область уменьшается в размерах за счёт прогрессирующей атрофии.
Изменению подвергаются и клетки эндотелия микроциркуляторного русла, причём это происходит уже с первых минут ишемии. Клетки набухают, проницаемость их мембран увеличивается. Примерно через 6 часов наблюдается некроз отдельных клеток, живые клетки (как эндотелиальные, так и гладкомышечные) пролиферируют, причём максимум происходит на 2-е - 3-и сутки ишемии [77]. С 5-го - 7-го дня наблюдается также ангиогенез, обеспечиваемый ангиогенными факторами роста, молекулами адгезии, в первую очередь интегринами. В целом это благоприятный признак, прогнозирующий улучшение восстановительных процессов в тканях мозга [78], [79].
1.2.3. Глутамат-кальциевый каскад
Глутамат-кальциевый каскад - это процесс массивного выброса возбуждающих нейромедиаторов глутамата и аспартата и накопления ионов кальция внутри клетки [80], [81], [82], [83], [84]. Он инициируется энергетическим дефицитом, возникающим при ишемии, а его следствием является повреждение нейронов и глиальных клеток. Условно выделяют три этапа глутамат-кальциевого каскада, соотносящиеся с этапами ишемического инсульта: индукцию, амплификацию и экспрессию.
Индукция. Локальное нарушение кровотока приводит к ограничению вплоть до полного прекращения доставки в мозг необходимых ему субстратов, в частности, кислорода и глюкозы. Это закономерно приводит к падению уровня АТФ, а также ГТФ и др. В свою очередь, это приводит к нарушению энергозависимого транспорта ионов [85], [86], деполяризации мембран нейронов и глиальных клеток, следствием чего является избыточное высвобождение глутамата, являющегося медиатором возбуждения. Глутамат накапливается в межклеточном пространстве, что ведёт к активации NMDA-рецепторов [87]. Вследствие этого открываются ионные каналы и в клетку неконтролируемо проникают ионы натрия и кальция, а также хлорид-анионы и вода [88], [89], [90]. В связи с проникновением воды клетка набухает и в конечном счёте разрушается. Пока этого не произошло, наблюдающееся нарушение функции нейронов является обратимым. Параллельно с обозначенными процессами происходит активация глутаматных рецепторов третьего типа, чувствительных к изменению обмена веществ [91], [92], [93], [94]. При этом происходит стимуляция образования диацилглицерина и инозитол-1,4,5-трифосфата, вступающих в действие на этапе амплификации.
Амплификация. Инозитол-1,4,5-трифосфат стимулирует высвобождение кальция из внутриклеточных депо (гладкого эндоплазматического ретикулума) [95], [96], приводя к увеличению его концентрации внутри клетки. Кроме того, кальций продолжает поступать в клетку через потенциалзависимые кальциевые каналы, открывшиеся в результате изменения мембранного потенциала, и в результате работы трансмембранного натрий-кальциевого переносчика, активирующегося избытком ионов натрия. Избыток ионов кальция и диацилглицерина активирует ферменты, модифицирующие мембранные белки, значительную долю которых составляют рецепторы, в том числе рецепторы глутамата. В результате этого возрастает чувствительность клетки к возбуждающим сигналам, нарастает деполяризация мембран за счёт проникновения в клетку катионов, в том числе Ca2+, в связи с чем высвобождение глутамата продолжается и увеличивается. Возбуждение соседних нейронов усугубляется [97]. Изменения нейронов на этом этапе ещё обратимы.
Экспрессия. Гиперактивация рецепторов глутамата приводит к нарушению ионного гомеостаза, накоплению внутри клетки ионов кальция. В какой-то момент изменения становятся необратимыми и клетка погибает. Кальций связывается с внутриклеточным белком-рецептором кальмодулином, вызывающим активацию ряда ферментов – протеаз, фосфолипаз, циклооксигеназы, NO-синтазы. В результате их работы расщепляются белки, липиды и нуклеиновые кислоты (в первую очередь РНК) [98], [99]. В частности, действие фосфолипазы разрушает плазмалемму и внутриклеточные мембраны. При распаде фосфолипидов может образовываться арахидоновая кислота, вызывающая появление в клетке активного кислорода [100], [101], [102], [103]. Происходит реакция перекисного окисления липидов, что усугубляет процесс распада мембран. Это приводит к образованию фактора активации тромбоцитов [104], итогом работы которого в конечном счете становится нарушение микроциркуляции, ещё более усиливающее ишемический процесс.
1.2.4. Реакция генома на ишемию
Ответная реакция организма на острую энергетическую и кислородную (что взаимосвязано) недостаточность представляет собой упорядоченную совокупность срочных и отложенных процессов компенсации разрушительных процессов. В нейронах запускается каскадный механизм поэтапной активации экспрессии генов [105], [106].
На снижение мозгового кровотока нервная ткань в первую очередь отвечает снижением синтеза белка и мРНК (что в конце концов проявляется в компактизации хроматина и изменении формы клеток). После чего включается неспецифическая реакция генома – синтезируются мРНК генов раннего реагирования, продукты которых могут участвовать в регуляции экспрессии генов позднего действия (каскадный механизм). Это происходит уже в первые минуты патологического процесса и является ответом на нарушение процесса активного транспорта ионов, вызванного недостатком АТФ в условиях недостатка кислорода и глюкозы, на изменение мембранного потенциала, вызванного в свою очередь нарушением процесса ионного транспорта, и на выброс глутамата как медиатора возбуждения [107]. Экспрессия генов раннего реагирования чаще всего приводит к синтезу ДНК-связанных белков – транскрипционных факторов, вызывающих экспрессию большого числа генов через активацию промоторных элементов [108]. Ряд генов раннего реагирования кодирует молекулы, вовлечённые в биохимические каскады сигнальных путей: протеинфосфотазы, G-белки, циклооксигеназу и др. [109]. Низкие концентрации кислорода индуцируют активацию специфического транскрипционного фактора HIF-1 [110], выполняющего функцию регуляции гомеостаза кислорода в клетке, для которого идентифицировано более 60 генов-мишеней, продукты которых способствуют улучшению доставки кислорода и метаболической адаптации. Это в частности фактор роста эндотелия сосудов VEGF и эритропоэтин. В результате их работы запускаются процессы ангиогенеза и неоваскуляризации, а также эритропоэз. В результате этого увеличивается поступление кислорода в клетку [111]. Также мишенями HIF-1 являются гены, кодирующие микроРНК miR-210 и miR-373, регулирующие экспрессию генов репарации ДНК [112], и гены гистоновых деметилаз [113]. Таким образом, уже с первых часов гипоксии клеточный ответ является скоординированным и затрагивает разные уровни регуляции экспрессии генов.
Также активируется экспрессия генов белков теплового шока – стресс-белков, или шаперонов, которые ответственны за сворачивание (фолдинг) белка и формирование толерантности к ишемии. Транскрипционные факторы этих генов активируются путём фосфорилирования вследствие увеличения концентрации кальция внутри клетки, свободнорадикальных реакций, активации протеазных ингибиторов и тирозинкиназ. Основная причина запуска синтеза шаперонов – дефицит АТФ, возникающий вследствие недостаточного поступления кислорода и глюкозы в клетку [114], [115].
Следующая волна экспрессии генов, формирующаяся под влиянием ишемии, связана с генами, кодирующими регуляторы процессов, участвующих в механизмах отсроченной гибели клеток. Запускается синтез цитокинов и хемокинов, индукцирующих локальные воспалительные процессы в очаге ишемии, увеличение сосудистой проницаемости и гематоэнцефалического барьера [116]. Также запускаются гены NO-синтетазы и циклооксигеназы-2. Всё это происходит в нейтрофилах, глиальных клетках и клетках эндотелия сосудов и достигает максимума через 12 часов после окклюзии средней мозговой артерии [117], [118]. Кроме того, экспрессируются гены, кодирующие продукты, участвующие в таких процессах, как апоптоз, выживание клеток, нейропластичность.
Итак, церебральная ишемия вызывает серьёзные изменения в экспрессии генома и запускает сложную разветвлённую систему генетических программ, определяющих по какому пути – гибели или выживания – пойдут клетки.
1.2.5. Комплексины и их возможное участие в глутаматной эксайтотоксичности
Комплексины – это семейство специфических эволюционно консервативных белков цитоплазмы нейронов, связывающихся с рецептором белка а-SNAP – белковым комплексом SNARE, состоящим из синтаксина, синаптобревина и SNAP-25 и обеспечивающим высвобождение медиаторов из синаптических пузырьков в синаптическую щель. Известно, что существует два типа комплексинов – CPLX1 и CPLX2. Первый представлен в тормозящих (ингибирующих) синапсах, второй – в возбуждающих [119], [120]. Это сравнительно небольшие по размеру (молекулярная масса около 19000 Да) белки цитозоля нейронов. Комплексины богаты остатками глутаминовой кислоты и лизина [121]. В основном локализуются в пресинаптических окончаниях нейронов млекопитающих. Белковая молекула состоит из 4 функциональных частей:
- N-концевой домен,
- вспомогательная спираль,
- центральная спираль,
- С-концевой домен [122].
Комплексины выполняют двойную функцию: они могут действовать и как активаторы, и как ингибиторы экзоцитоза в зависимости от физиологического состояния клетки. Ингибирование слияния мембран необходимо для предотвращения самопроизвольного экзоцитоза в синапсе [123], [124]. Последние данные поддерживают версию, что синаптотагмин играет роль в конформационных изменениях SNARE при изменении концентрации кальция [123]. Связывание кальция с синаптотагмином провоцирует изменение конформации и вытеснение комплексина, после чего мембрана везикулы сливается с пресинаптической мембраной и происходит экзоцитоз [125]. При низкой концентрации кальция экзоцитоз-ингибирующий эффект комплексина выражен сильнее. Он снижается посредством синаптотагмина при растущей концентрации кальция с переводом синаптотагмина в активное состояние [123].
Исследования, проведённые Н.М. Раевской и др., обнаружили у человека два варианта транскриптов гена Cplx2, различающиеся 5’- концевыми участками (CPLX_v1 и CPLX_v2). Они имеют высокое сходство с аналогичными транскриптами у крыс (Cplx_v1 и Cplx_v2) [126]. Этот случай не уникальный, он известен для многих генов, причём различные транскрипты одного и того же гена часто являются тканеспецифичными либо появляются в одной и той же ткани и клетке в зависимости от стадии клеточного цикла.
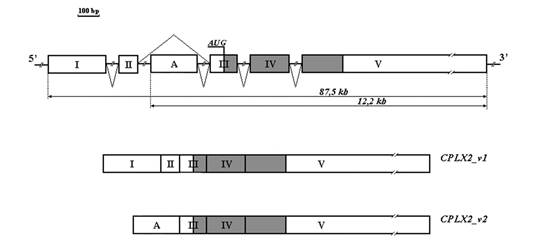
Рисунок 1.1. Структурная организация гена CPLX2 человека и его транскриптов
Факторы, определяющие транскрипцию по одному или по другому типу, до конца не выяснены. Вероятно, роль в «выборе» того или иного продукта играют альтернативные промоторы и взаимодействующие с ними факторы, что определяется потребностью клетки в том или ином продукте.
Исследования экспрессии гена Cplx2 также показали двустороннюю роль комплексинов в механизме экзоцитоза. Так, например, на культуре клеток РС12 было обнаружено, что Cplx2 стимулирует экзоцитоз при низком экспрессионном уровне и ингибирует при более высоком [127]. Изменение экспрессии гена этого белка при ишемии мало изучено, хотя известно, что важнейшие звено в механизме развертывания ишемического повреждения ткани мозга – глутаматная эксайтотоксичность – частично обеспечивается высвобождением глутамата в составе синаптических везикул. Также показано, что Cplx2 активно экспрессируется не только в нейронах, но и в астроцитах, которые принимают важное участие в регуляции оборота глутамата [128]. Таким образом, белок CPLX2 может принимать важное участие в глутаматной эксайтотоксичности. Например, было показано, что увеличение экспрессии Cplx2 приводит к нарушениям высвобождения глутамата с последующими патологическими изменениями [129], [130]. Также следует отметить, что функции комплексинов связаны с регуляцией экзоцитоза не только глутамата, но и других важных медиаторов и трофических факторов.
1. 3. Нейропротекция при ишемии
Лечение ишемического инсульта включает в себя два основных компонента – улучшение перфузии мозга и нейропротекцию. При этом выделяют первичную нейропротекцию, направленную на прерывание быстрых реакций самых ранних процессов ишемического каскада в пределах «терапевтического окна», и вторичную нейропротекцию, которая направлена на уменьшение выраженности отдалённых последствий ишемии.
Первичная нейропротекция основана на применении препаратов – антагонистов глутаматных NMDA и AMPA рецепторов, в которые встроены агонист-зависимые ионные каналы, играющие значительную роль в повреждении тканей мозга при ишемии. Антагонисты могут быть конкурентными (селфотел, элипродил и др.), блокирующими глутамат-распознающие сайты рецепторов и уменьшающими таким образом поток ионов кальция в клетку, и неконкурентными (фенциклидин, кетамин, дизолципин, декстрометорфан, декстрорфан, церестат, ремацемид, магний).
Также стратегией первичной нейропротекции является торможение синтеза и пресинаптического высвобождения глутамата, приводящее к уменьшению выраженности постсинаптической эксайтотоксичности. Препараты этого ряда – BW-619C89, пропентофиллин, фенитоин, фосфенитоин, лубелузол.
Кроме того, для уменьшения глутаматной эксайтотоксичности проводится устранение нейротрансмиттерного дисбаланса путём активации тормозных систем. В данном случае наиболее часто применяются ГАМК и глицин.
Вторичная нейропротекция направлена на прерывание отсроченных механизмов смерти клеток: избыточного синтеза NO и оксидантного стресса, активации микроглии и дисбаланса цитокинов, иммунных сдвигов, локального воспаления, нарушения микроциркуляции и ГЭБ, трофических нарушений и апоптоза. Важным направлением вторичной нейропротекции является антиоксидантная терапия [131].
1.3.1 Семакс и PGP
Последствия церебральной ишемии являются основанием для применения модулирующих веществ, способных устранять нарушения как локального, так и системного характера на молекулярном уровне - способных к нейропротекции, а также восстанавливающих микроциркуляцию в тканях мозга, стимулирующих репаративные процессы, предотвращающих вторичные повреждения.
При вторичной нейропротекции исключительно важную роль могут играть нейропептиды – регуляторы деятельности ЦНС. В последнее время большое внимание исследователей направлено на изучение фармакологических свойств меланокортинов – большого семейства нейропептидов, образованных из общего предшественника – молекулы проопиомеланокортина и включающего в себя группу меланоцитстимулирующих гормонов, АКТГ, а также их фрагменты. Примером такого нейропептида является семакс – синтетический пептид (метионил-глутамил-гистидил-фенилаланил-пролил-глицил-пролин), обладающий нейрозащитными, психостимулирующими и антигипоксическими свойствами. Он представляет собой модифицированный фрагмент АКТГ(4-7), Met-Glu-His-Phe). С конца 1970-х годов Институтом молекулярной генетики совместно с МГУ им. Ломоносова производились разработки препарата, способного оказывать вышеуказанный эффект. Поскольку большинство экзо- и эндопептидаз не расщепляют последовательности, обогащённые пролиновыми остатками, был синтезирован ряд аналогов АКТГ с участками, обогащёнными пролиновыми остатками с C-конца пептида. В результате аналог, содержащий концевой мотив PGP, наиболее устойчив, что приводит к значительному усилению эффекта и его пролонгированию [132].
Препарат «Семакс» оказывает многофакторное нейропротекторное действие: в частности, он активирует синтез нейротрофинов – регуляторов роста и дифференцировки нервной ткани, усиливает внимание, улучшает память, способствует адаптации организма к гипоксии, церебральной ишемии, наркозу и т.д. [133], [134]. Препарат вводится в низких дозах, интраназально, в незначительном количестве проникает через гематоэнцефалический барьер [135], при попадании в ткани распадается до отдельных аминокислот. Концевой трипептид PGP несколько задерживает этот процесс во времени, продлевая эффект препарата. При отщеплении метионина образуется довольно стабильный фрагмент Glu-His-Phe-Pro-Gly-Pro [136], который, тем не менее, через некоторое время деградирует до аминокислот. Наиболее стабилен С-концевой трипептид PGP.
Опыты на культуре нервной ткани показали выраженное трофотропное действие семакса на нейроны холинергической группы, в том числе в условиях недостатка кислорода и глюкозы.
Установлено, что при использовании препарата баланс пептидэргических систем мозга сдвигается в сторону преобладания противовоспалительных и защитных факторов над факторами, поддерживающему воспалительные реакции. Семакс влияет на процессы синтеза оксида азота (в сторону его уменьшения), состояние рецепторов глутамата и баланс нейротрансмиттеров.
PGP (пролил-глицил-пролин), образующийся при деградации семакса, обнаруживается в плазме крови спустя 3-5 часов после введения препарата в концентрации не выше 2 – 5% от количества введённого гептапептида. В дальнейшем он ферментативно распадается с образованием дипептидов PG и GP [137]. Пролин-содержащие пептиды воздействуют на ряд процессов в организме, в частности, они обладают протекторным действием на слизистую оболочку желудка. К сожалению, молекулярные механизмы действия семакса остаются до конца не выясненными. Ранее было высказано предположение, что нейропротекторные эффекты семакса связаны с увеличением экспрессии нейротрофинов [138]. Кроме того, в ряде работ были показаны и другие аспекты действия семакса: иммуномодуляция, торможение воспалительных реакций, торможение синтеза оксида азота и реакций оксидативного стресса [139], [140].
Дата добавления: 2018-10-25; просмотров: 2859; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!