Фазовые равновесия и фазовые превращения при отверждении
РОССИЙСКОЙ ФЕДЕРАЦИИ
ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ
ОБРАЗОВАТЕЛЬНОЕ
УЧРЕЖДЕНИЕ ВЫСШЕГО ОБРАЗОВАНИЯ
«МОСКОВСКИЙ АВИАЦИОННЫЙ ИНСТИТУТ
Национальный исследовательский университет)»

Институт материаловедения и технологий материалов
Кафедра «Технология композиционных материалов, конструкций
И микросистем»
КУРСОВАЯ РАБОТА
По дисциплине: «Современные термодинамические подходы в материаловедении»
На тему: Фазовые состояния и превращения отверждающихся олигомер-полимерных систем
Студент гр. Т110-106М-19 /Сосков Д.Ю./
(подпись)
Руководитель __________________________________________________/Бабаевский П.Г./
(подпись)
МОСКВА – 20 20
Содержание
| Лист | |
| ВВЕДЕНИЕ | 3 |
| 1. Фазовые равновесия и фазовые состояния исходных отверждающихся олигомер-полимерных систем | 4 |
| 2. Фазовые равновесия и фазовые превращения при отверждении | 10 |
| 3. Фазовые равновесия и фазовые превращения в отвержденном состоянии, фазовая морфология | 16 |
| ЗАКЛЮЧЕНИЕ | 25 |
| СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ | 26 |
ВВЕДЕНИЕ
|
|
|
В результате анализа исследований процессов структурирования, протекающих в гетероцепной макромолекулярной среде. Преимущественно методами ядерного магнитного резонанса (ЯМР) показано, что они имеют как минимум две особенности:
во-первых, происходит преобразование механизмов и сдвиг равновесия у реакций межцепного обмена, сопутствующих процессам структурирования олигомеров и полимеров;
во-вторых, упаковка макромолекулярного ансамбля по мере его структурирования сопровождается самопроизвольной ориентацией макроцепей (их самоорганизацией).
В этой связи рассмотрена макросегментальная модель формирования и строения аморфной фазы полимера, учитывающая вышеописанные особенности существенно неравновесных процессов отверждения гетероцепных олигомеров. В рамках той же модели оцениваются результаты фазовых превращений в смесях сополимерных полиолефинов, модифицированных малыми добавками олигомера.
1.
Фазовые равновесия и фазовые состояния исходных отверждающихся олигомер-полимерных систем
Фазовые равновесия - равновесия в гетерогенных системах, в которых не происходит химического взаимодействия, а имеет место лишь переход компонентов из одной фазы в другую или другие.
|
|
|
Фазовая диаграмма вещества наглядно показывает области температур и давлений, в которых термодинамически устойчивы разные фазы данного вещества. Не все фазы полностью отделены друг от друга линией фазового перехода. В некоторых случаях эта линия может обрываться, оканчиваясь критической точкой. В этом случае возможен постепенный, а не скачкообразный переход из одной фазы в другую, в обход линии фазовых переходов Набор термодинамических фаз вещества обычно значительно богаче набора агрегатных состояний, т. е. одно и то же агрегатное состояние вещества может находиться в различных термодинамических фазах.
Диаграммы состояния строят для условий равновесия или условий, достаточно близких к ним. Они дают наглядное представление о процессах, происходящих в системах при нагревании и охлаждении, что может быть использовано при оценке физико-химических, механических и технологических свойств систем и позволяет рационально подойти к выбору материалов для изготовления изделий.
Диаграмма, выражающая зависимость состояния гетерогенной системы от внешних условий, называется диаграммой состояния или фазовой диаграммой.
|
|
|
В однокомпонентной системе число независимых компонентов K=1 и правило фаз Гиббса имеет вид:
C = K – Ф + 2 = 1 – Ф +2 = 3 – Ф
В этом случае в качестве переменных параметров взяты p и T. Поскольку число степеней свободы С ≥ 0, то 3 – Ф ≥ 0, откуда Ф ≤ 3, т.е. число равновесных фаз не должно быть больше трёх, поэтому могут существовать одно-, двух- и трехфазные однокомпонентные системы. Все эти фазы состоят из одного и того же вещества в различных агрегатных состояниях (газ, жидкость, твердое) или в различных кристаллических модификациях. Диаграмма состояния гетерогенной системы представляет собой графическое выражение связи между p, T и составом отдельных фаз.
В процессе отверждения олигомер-полимерной системы химические процессы резко тормозятся. Вследствие этого формируются недоотвержденные стеклообразные материалы, содержащие определенное количество непрореагировавших функциональных групп. Время перехода в стеклообразное состояние реагирующей системы можно оценить по так называемым TTT диаграммам (time-temperature-transformation), представляющим собой корреляции между временем, соответствующим различным изменениям в системе (например, гелеобразованию или стеклованию), и постоянной температурой отверждения, при которой это время было получено. Однако, ТТТ-диаграммы могут дать информацию не только о времени стеклования, но и о температуре, выше которой следует нагреть систему для достижения близкой к 100% конверсии, а также времени, необходимого для этого. Другими словами, по ТТТ-диаграммам можно обоснованно прогнозировать режим доотверждения.
|
|
|
В качестве критерия окончания первой стадии (собственно отверждения), можно использовать время, соответствующее достижению как стеклования, так и гелеобразования и иных состояний системы. В связи с этим возникает вопрос: влияет ли выбор указанного критерия на степень отверждения и свойства полностью отвержденных систем, или нет. Таким образом, целью настоящей работы было исследование механических свойств полностью отвержденных по различным двухстадийным режимам олигомер-полимерных систем, и установление факта влияния режима отверждения на эти свойства.

Рисунок 1 - Типичные температурные зависимости модуля упругости (а) и модуля потерь (б) эпокси-аминных полимеров с температурой предотверждения 1 – 8, 2 – 22 и 3 – 60°C
За время начала реакции принимали момент добавления отвердителя в индивидуальный эпоксидный олигомер. Отверждение проводили в две стадии. На первой (предотверждение) реакционноспособные композиции выдерживали при постоянных температурах 8±2, 20±1 и 60±1ºC до времени t1, при котором завершается перестройка надмолекулярной структуры исходной эпокси-аминной системы [6, 7]; времени tgel, соответствующего гелеобразованию, а также времени tg, соответствующего переходу в стеклообразное состояние. Время и температуру второй стадии (доотверждения) для получения предельно сшитой системы определяли по ранее полученным ТТТ-диаграммам [5]. Исследования проводили с использованием динамического механического анализатора DMA Q800 TA Instruments [8] в температурном диапазоне от 0 до +130 ºC при скорости нагрева w+ =3 ºC/мин в атмосфере сухого воздуха.
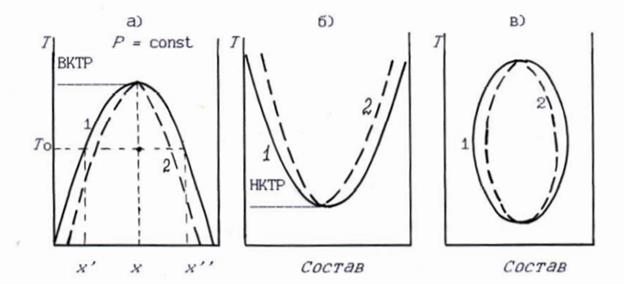
Рисунок 2 - Некоторые типы фазовых диаграмм двухкомпонентной некристаллизующейся системы: а - равновесие двух ограниченно смешивающихся жидкостей с верхней критической температурой расслоения (ВКТР); б - то же с нижней критической температурой расслоения (НКТР); в - наиболее общий вид фазовой диаграммы некристаллизующихся веществ. 1 - бинодаль; 2 - спинодаль; x - раствор критического состава; x' и x" - составы сосуществующих фаз в системе состава x
На фазовых диаграммах в координатах температура - состав точки образуют граничную кривую равновесия фаз - бинодаль. Наиболее общая форма бинодали - замкнутая кривая (овал) – рис.2в. Точки внутри этого овала характеризуют температуры и составы, при которых раствор распадается на 2 фазы, является гетерогенным; вне этого овала система однофазна, гомогенна.
В подавляющем большинстве случаев наблюдается лишь часть овала. Нижняя часть овала часто не может быть реализована из-за кристаллизации или стеклования раствора, а верхняя часть - из-за достижения во многих случаях температуры кипения растворителя или деструкции компонентов раствора при давлении, поддерживаемом в эксперименте. Поэтому в реальных растворах бинодаль имеет форму незамкнутой кривой в первом случае с максимумом, во втором - с минимумом (рис. 2а и 2б). Если действуют оба ограничения, то реализуется только средняя часть овала, и бинодаль имеет форму двух независимых кривых без экстремумов, между которыми лежит область распада раствора на две фазы. Максимумы и минимумы на бинодали (так называемые критические точки) определяют верхнюю (ВКТР) и нижнюю (НКТР) критические температуры растворения и критические концентрации. В критической точке составы сосуществующих фаз совпадают, при всех других температурах (например при температуре То) - не совпадают, их значения x' и x'' для состава x ясны из рис. 2а.
В некоторых системах возможно сближение или даже перекрытие верхней бинодали и нижней бинодали, отчего область полного смешения уменьшается или даже исчезает совсем для некоторого интервала составов.
Данные фазовые диаграммы систем с некристаллизующимися компонентами называют фазовыми диаграммами "аморфного равновесия".
Особенность систем аморфный характерен резкий сдвиг ВКТР и НКТР в сторону полимера. Такой сдвиг понятен, так как кинетическая подвижность больших молекул полимера очень мала. Концентрация полимера в фазе I (раствор полимера в жидкости) может составлять десятые и сотые доли процента или даже тысячные доли процента у очень высокомолекулярных полимеров, а для предельно жесткой цепи большой молекулярной массы должна быть практически равна нулю. Она отличается от нуля из-за гибкости цепей и наличия в полимере низкомолекулярных фракций.
Вторая особенность диаграмм - кривая фазового равновесия (бинодаль) фактически представляет собой не одну линию, а систему кривых, отвечающих набору фракций разной молекулярной массы. Это более характерно для левой части кривой аморфного расслоения. При построении диаграмм обычно не принимают во внимание полидисперсность полимера по молекулярной массе.
Следующая особенность диаграмм вытекает из неоднородности химического строения макромолекул. В пределах одной макромолекулы могут отличаться по строению мономерные звенья (в статистических сополимерах, например). Мономерные звенья могут соединяться по типу "голова к голове" или "голова к хвосту", боковые ответвления иметь разную длину, макромолекулы могут участвовать в образовании трехмерных сшитых структур.
Еще одна особенность фазовых диаграмм систем с участием полимеров состоит в том, что равновесие в таких системах очень сильно зависит от введения небольших количеств химически активных примесей - полифункциональных веществ, которые могут одновременно реагировать с двумя функциональными группами соседних макромолекул, сшивать их и тем самым резко смещать фазовое равновесие вплоть до распада однофазного гомогенного раствора на две фазы с образованием студня (геля) или рыхлого осадка полимера.
Характерным для полимерных систем является сочетание различных типов фазового равновесия: кристаллического с аморфным, аморфного с жидкокристаллическим, кристаллического с жидкокристаллическим.
Принципиально можно выделить два случая:
1. Установление одного типа равновесия до того, как установилось другое равновесие;
2. За пределами области одного типа фазового равновесия возможен другой тип распада на фазы. Конкретно это относится к системам, у которых выше или ниже кривой ликвидуса компоненты оказываются ограниченно совместимы.
Такими системами являются, например, смеси поли N,N'-себацоилпиперазин - дифениловый эфир и поливинилхлорид - бутилбензилфталат.
Фазовые равновесия и фазовые превращения при отверждении
Процессы структурирования неотделимы от процессов сеткообразования и фактически являются их заключительной стадией, им присущи определенные особенности, которые можно рассматривать с различных точек зрения:
· как золь-гель переход: c точки зрения коллоидной химии, реологии и морфологии, структурирование является конечным результатом преобразования относительно гомогенной вязкотекучей среды в гетерогенную нерастворимую и неплавкую массу;
· как сеткообразование: с позиций макромолекулярной химии и топологии структурирование – это следствие преобразования ансамбля макромолекул повышенной (>2) функциональности (линейных и(или) разветвленных) в сетчатые ((макро)циклические) структуры различного масштаба, то есть представляет собой масштабный переход от микро- к макроциклическому строению ММА;
· как переход жидкость-твердое тело: если исходить из начального и конечного физического (агрегатного) состояния макромолекулярного ансамбля, то структурирование предстает как конечный результат «химической закалки» или подавления трансляционной молекулярной подвижности путем образования дополнительных межмолекулярных связей (преимущественно химических) и соответственно возрастания среднего уровня (фона) межмолекулярных взаимодействий;
· как химически индуцированный фазовый переход: если рассматривать образующиеся гетерогенные множества частиц (условно золь- и гель-фракцию) в качестве различных термодинамических фаз, то структурирование – это завершающая стадия формирования (фиксации) границ между ними;
· как переход перколяции: если анализировать процесс с точки зрения скейлингового подхода, то структурирование оказывается масштабным переходом от локальной к макроскопической связности полимерных фрагментов, неоднородно распределенных по объему аморфной фазы сетчатого полимера.
При анализе химически индуцированных переходов основное внимание уделяется роли химических связей, формирующихся по ходу процесса отверждения. Необходимо также отметить, что последние три подхода можно рассматривать и как различные аспекты термодинамического метода анализа процессов структурирования (гелеобразования) сетчатых полимеров, основы и многие акценты которого закономерно смещаются в сторону неравновесной термодинамики, рассматривающей, в частности, и явления самоорганизации. При этом фрактальный подход к оценке результатов структурирования в определенной степени является симбиозом, дополнением и развитием коллоидно-топологических представлений, хотя и не сводится к ним непосредственно.
С позиций химии сеткообразование можно охарактеризовать как метод наращивания молекулярной массы полимеров или олигомеров с повышенной функциональностью (f>2) до максимально возможного уровня.
С точки зрения физики, сеткообразование – фазовоагрегатный переход, совершаемый макромолекулярным ансамблем из жидкоподобного (вязкотекучего) состояния в твердоподобное (высокоэластическое или стеклообразное) состояние под воздействием химических превращений (процесс «отверждения»). При этом возрастание энергии межмолекулярных взаимодействий и плотности молекулярной упаковки происходит не в результате варьирования внешних условий (температуры, давления и т.д.), а вследствие изменения концентрации, размеров и строения (в особенности топологии и упаковки) частиц, образующих исходный ММА (мономерный, олигомерный или полимерный).
По этой причине явление химического отверждения («химическую закалку») нельзя представлять как результат простого переохлаждения олигомерной жидкости, т.е. как следствие переноса (сдвига) функции ее состояния только по температурной шкале. В этой связи гелеобразование можно трактовать как необратимый агрегатный переход всего макромолекулярного ансамбля, вызываемый химически индуцированными фазовыми превращениями в пределах отдельных подансамблей. По-видимому, именно смена масштаба исходно наноразмерной олигомерной частицы по мере ее роста первоначально приводит к соответствующему переходу от агрегатного (в рамках олигомерного ансамбля) к фазово-агрегатному (коллоидному) состоянию (организации) макромолекулярного ансамбля с дальнейшим усреднением состояний отдельных его подансамблей в процессе химического сшивания или охлаждения вплоть до формирования единого агрегатного состояния всей макромолекулярной среды.
На фоне и в результате нарастающих в области золь-гель перехода межцепных взаимодействий физической природы, изменений в величине и механизме гибкости структурирующихся молекул, а также плотности их упаковки, создаются предпосылки для возбуждения гетероатомов цепи активными протонами и(или) ионами вплоть до формирования реакционноспособных аддуктов с их участием и соответственно роста химической активности макромолекулярной среды. При этом резко возрастает значение возникающих по ходу процесса или ранее незначительных конформационных и энергетических факторов, например, химических и физических проявлений «эффекта цепи» (смена механизма гибкости отдельных участков или цепей в целом, интенсивности и направления переходов типа «клубок-глобула», изменение вклада побочных реакций, в частности, циклизации, и т.п.). Одновременно усиливается роль комплексных физикохимических (хемосорбционных и т.п.) взаимодействий, что в совокупности меняет механизмы и(или) направления процессов и химической, и физической природы (вплоть до воздействия на уровень термодинамической совместимости олигомеров и(или) соответствующих полимеров).
В этом процессе могут участвовать и любые внешние или внутренние химически активные источники (вплоть до примесей разнообразной природы). Дальнейшая эволюция подобных аддуктов в почти «твердотельной» среде способствует выделению низкомолекулярных соединений и постепенному сдвигу равновесия межцепных реакций в сторону формирования зольфракции. Этот результат связан с ростом вязкости композиции, в результате чего растет и вероятность стерического обрыва ранее замкнутых кинетических цепочек с участием гетероатомов. Последнее приводит к постепенному затуханию межцепного обмена и соответственно прекращению локальной деструкции полимерных цепей (по крайней мере вследствие межцепных взаимодействий). Поэтому одной из основных химических особенностей процесса структурирования в гетероцепной среде является изменение не только механизмов, но и природы межцепных взаимодействий, которые по мере структурирования преобразуются из равновесных (обратимых) в неравновесные (необратимые).
При этом именно в процессах вулканизации (отверждения), и в особенности на стадии структурирования, реакционноспособных олигомеров в блоке проявляется тесная взаимосвязь явлений химической и физической природы, которая отражается и на геометрии ММА. Это обусловлено двойственным статусом макромолекул в среде себе подобных, когда каждый элемент макромолекулярного ансамбля выполняет функции и среды, и реагента одновременно [9,10]. Поэтому любая реакционноспособная макромолекулярная среда не только активно влияет на скорость и механизмы протекающих в ней химических реакций и физических процессов, но и испытывает встречное воздействие со стороны присутствующих в ней компонентов. Взаимодействие среды и реагентов становится особенно значительным в области золь-гель-перехода (на стадии структурирования), когда неоднородность ММА максимальна.
Реакционноспособные олигомеры являются не только широко распространенным сырьем для получения сетчатых полимеров, но и естественным исходным материалом для моделирования глубоких стадий процессов структурообразования, особенно в условиях, аналогичных тем, что характерны для технологий получения связующих и герметизирующих материалов (невысокие температуры и(или) значительная вязкость реакционной среды). Те же условия близко соответствуют условиям, в которых протекают золь-гель переходы независимо от их механизмов и(или) параметров отверждения, что позволяет рассматривать исследования процессов структурирования олигомерсодержащих композиций в качестве модельных для любых химически индуцированных процессов структурирования с участием высокомолекулярных соединений.
Образование сшивок макромолекул в полимере резко изменяет динамические механические свойства выше температуры стеклования. Так, при отверждении фенолоформальдегидных или эпоксидных смол динамический модуль тем больше, а пик механических потерь ниже и шире, чем больше количество отвердителя (рис. 3). При этом максимум механических потерь заметно смещается в сторону более высоких температур, а модуль медленнее изменяется с температурой. При очень высокой частоте сшивок температура стеклования либо исчезает совсем, либо становится выше температуры деструкции полимера, и максимума механических потерь не наблюдается (см. кривую 3 на рис. 3). Считается, что расширение области релаксационного перехода с увеличением частоты узлов сетки связано с увеличением ширины распределения молекулярной массы цепей между узлами сетки или появлением каких-либо других неравномерностей структуры сетки.

Рисунок 3 - Температурные зависимости динамических механических свойств новолачной феноло-формальдегидной смолы, отверждаемой разным количеством уротропина в %: 1 - 2,0; 2 - 4,0; 3 - 10,0
В некоторых термореактивных композициях неравномерность структуры сетки может приводить к появлению двух фаз. Двухфазная структура представляет собой микрогелевые частицы, распределенные в густосетчатой матрице и химически связанные с ней. Микрогелевые частицы могут зарождаться в активных центрах, где наблюдается избыток сшивающего агента или катализатора реакции отверждения. В таких двухфазных системах проявляется раздвоение пика механических потерь. Если процесс отверждения полимера с образованием густосетчатой структуры идет при температурах ниже температуры стеклования, то реакция протекает очень медленно и очень длительное время. Она может ускориться в процессе испытания полимера на механические потери, поэтому картина динамических механических свойств становится очень сложной и противоречивой, плохо воспроизводится.
3. Фазовые равновесия и фазовые превращения в отвержденном состоянии, фазовая морфология
Результаты морфологических исследований аморфных образований в высокомолекулярных средах будут давать хотя и очень важную, но лишь косвенную информацию о фазовой организации полимерных материалов. Это связано с тем обстоятельством, что далеко не каждая из визуально наблюдаемых особенностей «надмолекулярных» неоднородностей будет непосредственно связана со строением отдельного (единичного) «макросегмента», а относительно низкочастотные динамические свойства подобных образований вряд ли проявляются в потоке светового облучения.
Структурирование в сеткообразующих композициях сопровождается определенными изменениями в строении и упаковке макромолекулярного ансамбля. Но для наиболее эффективной реализации потенциальных возможностей межмолекулярных взаимодействий и соответственно для достижения максимальной плотности молекулярной сетки требуется обеспечить оптимальный характер и плотность упаковки макромолекулярного ансамбля по всему объему полимера.
Особенности морфологии системы способны охарактеризовать ее дисперсность в макроскопическом масштабе, а также ЯМРрелаксометрия, позволяющая описывать структурно-динамические переходы, соответствующие более мелким масштабам – вплоть до наноуровня.
Морфология слабосшитых полимеров (клубки, мицеллы, фибриллы, домены и т.п.) характеризуется явно выраженным внутренним строением, то о глобулах гибкоцепных синтетических полимеров ничего, кроме их сферической формы и повышенной плотности по сравнению с остальным объемом аморфной фазы, практически неизвестно.
Определенную информацию о характере макроскопической связности исследуемых смесей можно извлечь из рис. 4-5. Исходя из масштаба наблюдаемых неоднородностей и близости значений молекулярных масс полимеров, можно предположить, что морфология исходных компонентов полимерных смесей (рис.4) заметно различается по своей природе.

Рисунок 4 - Морфология смесей хлорсульфированного полиэтилена (ХСПЭ) (а) и этиленпропиленового каучука тройного (СКЭПТ) (б) по данным электронной микроскопии (увеличение 10000х).
У ХСПЭ (рис.4а) она преимущественно связана со структурными образованиями макромолекулярного (микрометрового) масштаба, хорошо различимыми в поле зрения выбранного масштаба оптического увеличения, то подавляющая часть площади скола образца СКЭПТ (рис.4б) представляет собой относительно гладкую поверхность.
Это означает, что с учетом кратности увеличения микроскопа масштаб структурной и(или) оптической неоднородности этой поверхности приближается к нанометровому диапазону и поэтому может быть в значительной степени обусловлен сополимерным составом СКЭПТ. В ХСПЭ в дополнение к подобному гладкому «плато» (или основанию) и одновременно на его поверхности обнаруживается набор достаточно крупных полимолекулярных образований. Поэтому по сравнению со СКЭПТ морфология ХСПЭ более разномасштабная, т.е. и плотность упаковки макромолекул ХСПЭ со значительно большим разбросом (неоднородностью) по величине.
При избытке одного из компонентов в смеси внешне все соответствует ситуации, когда преобладающий по массе полимер образует непрерывную фазу, а оставшийся компонент – дискретную. Действительно, при минимальном содержании ХСПЭ в смеси (рис.5в) ее морфология определяется морфологией СКЭПТ, т.е. микрофазы ХСПЭ как бы «растворяются» в матрице СКЭПТ (предположительно на сополимерном уровне). Однако при обратном соотношении компонентов не наблюдается полной аналогии (рис.5а), соответствующей симметричной инверсии фаз: при избытке ХСПЭ сохраняются темные вкрапления, аналогичные тем, что наблюдаются при избытке СКЭПТ. При этом матрицы обоих полимеров как бы «накладываются» друг на друга, причем матрица СКЭПТ визуально заполняет и одновременно сглаживает пространство между полимолекулярными «долями» ХСПЭ и (или) участвует в их формировании. Тем самым она (в отличие от матрицы ХСПЭ) формирует смешанную фазу, одновременно включая в себя предположительно наиболее плотные (на рисунке – наиболее темные, сгусткообразные) дискретные фрагменты ХСПЭ.

Рисунок 5 - Морфология смесей ХСПЭ и СКЭПТ в зависимости от соотношения их компонентов: а - ХСПЭ/СКЭПТ 75/25 (3:1 мол.); б - ХСПЭ/СКЭПТ 50/50(1:1мол.); в - ХСПЭ/СКЭПТ 25/75(1:3 мол.)
Внешне (по формальным признакам) понятна и морфология равновесовой смеси полимеров (рис.5б): здесь наблюдается рифленая поверхность с темными дискретными впадинами, образованными преимущественно матрицей ХСПЭ, и более светлыми, непрерывными по своей топологии, возвышенностями, не содержащими специфических доменных образований и, повидимому, сформированными цепями СКЭПТ.
В первом приближении это выглядит как сосуществование двух равноправных непрерывных фаз, что позволяет ХСПЭ (полимеру с более плотной физической сеткой – энергией когезии) оставаться в рамках равновесовой композиции системообразующим компонентом (матрицей). Это подтверждается данными термомеханического и физико-механического анализа, согласно которым Тс и σр соответствующих смесей (с содержанием ХСПЭ >= 50 мас. ч.) определяются именно ХСПЭ.
Кроме того, подобные изображения (рис. 2б) известны как «полосатые структуры» и обычно свидетельствуют об абсолютной несовместимости полимеров [2]. Таким образом, смешение ХСПЭ и СКЭПТ, содержащего ЭНБ, при равновесовом соотношении компонентов приводит ко взаимному размежеванию полимеров.
При этом структурирование ММА сначала создает предпосылки для такого изменения механизма межцепных процессов, в рамках которого становится возможным сдвиг локального равновесия в сторону увеличения числа концевых цепей и доли (по крайней мере связанной) золь-фракции в системе. Затем оно способствует реализации потенциального сдвига равновесия в рамках нового механизма и затуханию любой химической активности по стерическим причинам. Поэтому любая диспропорция между реакциями локальной деструкции и структурирования, особенно в смеси термодинамически несовместимых компонентов и(или) при локальном избытке соединений с активными протонами (R-H), может стать дополнительным источником фазовых и структурных неоднородностей различного уровня - от деталей строения и компоновки отдельных участков полимерных цепей до природы связей и характера дефектности (упаковки) молекулярных сеток отверждённых олигомерных композиций.
При этом в процессе структурирования гетероцепных олигомеров не только преобразуются механизмы и сдвигается равновесие у межцепных процессов, но и в той или иной степени реализуется тенденция к упорядочиванию межузловых цепей по ходу изменения их длины и гибкости. В результате переход в полимерное состояние сопровождается возникновением особой («макромолекулярной») разновидности молекулярной подвижности и соответственно механизма ядерной магнитной релаксации в области температур и(или) молекулярных масс развитого сегментального (т.е. макросегментального) движения.
Основным источником отмеченных закономерностей является возрастание структурно-динамической неоднородности макромолекулярного ансамбля в области золь-гель перехода, которая, в том числе, приводит к изменению спектра молекулярных движений и механизмов перераспределения кинетической энергии и энтропии вдоль и поперек полимерных цепей. В результате создаются предпосылки для сдвига равновесия в ходе межцепных процессов и несоблюдения статистических закономерностей при формировании сетчатых структур.
Процессы структурирования в гетероцепной полимерной среде имеют как минимум две особенности: происходит преобразование механизмов и сдвиг равновесия у сопутствующих структурированию межцепных процессов; упаковка макромолекулярного ансамбля по мере его структурирования сопровождается упорядочиванием макроцепей.
При этом возможный механизм структурирования закономерно включает в себя стадию формирования макросегментов (подансамблей макромолекул), в рамках которых объединяются фрактальный и геометрический уровни полимерной структуры. Их сочетание не всегда проходит безболезненно для ММА и сопровождается распадом полимерных цепей, продукты которого вносят свой вклад как во фрактальный показатель (степень неоднородности и(или) дефектности) строения сетчатой структуры, так и в ее золь-фракцию.
Совокупное действие химических и физических факторов, возникающих в вязкой гетерогенной среде на глубоких стадиях процесса структурирования, приводит к ужесточению блоксополимерных цепей и относительному росту их «хрупкости», что способствует разрыву ослабленных гетеросвязей (в особенности расположенных на границах гибких и жестких блоков) и соответственно частичному распаду макромолекулярных цепей и формированию более плотной упаковки как гибких блоков, так и полимера в целом.
Данные закономерности можно также охарактеризовать как проявления тенденции к самоорганизации ММА по мере его структурирования (роста связности). Самоорганизация относится к категории самопроизвольных процессов с обратной связью, для которых характерно понижение энтропии системы, находящейся вдали от термодинамического равновесия или существующей преимущественно в волновой среде [4]. В последнем случае одним из признаков самоорганизации является преобразование параметров реакционной или молекулярной среды (например, полимерной цепи) с возникновением соответствующей обратной связи, обеспечивающей появление или рост упорядоченности структуры или движения её компонентов.
При этом максимально плотная упаковка достигается не только благодаря структурнодинамическим, но и структурно-химическим переходам, т.е. участию в процессах структурирования химического механизма перемешивания полимерных цепей и(или) сегрегации блоков различной природы, а также вследствие постепенного преобразования гетероатомных группировок в составе макромолекул в более устойчивые аналоги, содержащие меньшее число гетероатомов. Поскольку олигомерные молекулы в конденсированном состоянии одновременно выступают в двух «ипостасях»: реагентов и реакционной среды по отношению друг к другу, то есть любые их преобразования автоматически и одновременно становятся и преобразованиями реакционной среды, то отсюда любые, в особенности колебательные, процессы с обратной связью, сопутствующие подобному преобразованию, становятся проявлениями конкретного механизма самоорганизации (например, КРС, НМО, КП), конечным итогом и символом которого в данном случае являются макросегменты. При этом необходимо иметь в виду, что под «обратной связью» понимаются взаимодействия, по своей природе или масштабам не обязательно совпадающие с исследуемыми закономерностями, а механизм реализации обратной связи значительно упрощается именно в колебательной среде.
Самоорганизация макромолекул оказывается закономерным участником процесса структурирования сеткообразующих олигомерных композиций не только благодаря частичному сохранению в составе композита областей упорядоченности, сформированных в рамках исходных олигомерных структур [11,12] (что наиболее характерно для процесса отверждения олигоэфиракрилатных систем), но и вследствие значительной неравновесности макромолекулярного ансамбля в области золь-гель перехода. Последнее способствует как локальной деструкции полимерных цепей (которая особо характерна для эпоксиаминных и полисульфидных композитов), так и их взаимной ориентации (что более типично для полиуретановых систем).
Процессы самоорганизации следует рассматривать как фактор, способствующий формированию наиболее плотной, но в то же время и анизотропной упаковки макромолекулярных фрагментов данной топологии, прежде всего на молекулярном уровне. Это обстоятельство в зависимости от механизма упаковки фрагментов более крупного масштаба и соответственно вытекающей из него фрактальной структуры и подвижности макромолекул, может способствовать как ослаблению, так и усилению структурно-динамических неоднородностей, присущих всему объему материала, получаемого на основе олигомеров.
Различия в механизмах смешивания обусловлены разномасштабностью проявления фактора совместимости в различных композициях, что является следствием его «дуализма», т.е. зависимости ММА и соответственно совместимости полимеров минимум от двух структурнодинамических масштабов (сегментально-группового и макромолекулярного (фазового) или макросегментального).
Процесс сшивания макромолекул приводит не только к стабилизации фазовой структуры полимерных смесей, но и к повышению масштаба структурно-динамической неоднородности композиций.
Для полноценной оценки характера упаковки и межмолекулярных и(или) межфазных взаимодействий в полимерных системах первостепенного внимания требует анализ стерического (энтропийного) фактора, который в макромолекулярной среде проявляется в форме разнообразной и широкомасштабной конформационной структуры ее компонентов (от группового до макросегментального уровня).
Структурнодинамическая самоорганизация гибкоцепных макромолекул, включающая в себя элиминацию наиболее напряженных гетероатомных связей (в особенности в составе скелетных цепей макромолекул) является неотъемлемой компонентой полномасштабного процесса структурирования любого олигомерного ансамбля, в особенности, в присутствии больших количеств активных наполнителей, т.е. ориентирующих и(или) (хемо)сорбирующих добавок, способствующих дестабилизации любых, в особенности гетероатомных, полимерообразующих связей.
По мере формирования высокомолекулярных структур возрастает влияние реакционной среды, которая сама начинает принимать участие в процессах гелеобразования. Изменения физико-химических свойств реакционной среды отражаются и на механизмах протекающих превращений. Возникающая обратная связь между средой и промежуточными реагентами приводит не только к переменам в механизмах отдельных химических актов, но и к существенным изменениям в характере процесса структурообразования в целом, включая макроскопические последствия его действия. В результате процессы гелеобразования, которые сопровождаются четким разделением системы на золь– и гель–фракцию, постепенно преобразуются в процессы структурирования, которые предполагают по крайней мере частичное совмещение этих фракций (фаз) в рамках единого структурно-динамического образования. Формирование связной макроскопической структуры в пределах всех возможных масштабов и по всему объему молекулярной системы (т.е. ее макроскопическое «структурирование») предполагает включение в состав полимерного материала и низкомолекулярных, в особенности химически связанных, продуктов соответствующих превращений. Это способствует не только росту распределения по вязкости и гетерогенности формирующейся структуры, но и изменению химического потенциала молекулярной системы в целом, а также механизмов отдельных элементарных актов химических процессов в ее микрообъемах.
Поэтому в условиях физико-химической неоднородности и, одновременно, сильного взаимодействия, обеспечивающего относительное единство реакционной среды и реагентов, более адекватной «физико-химической» характеристикой наблюдаемых процессов структурирования может быть не столько величина pH системы, сколько особенности поведения активных атомов и(или) ионов водорода (протонов).
ЗАКЛЮЧЕНИЕ
Совокупное действие химических и физических факторов, возникающих в вязкой гетерогенной среде на глубоких стадиях процесса структурирования, приводит к ужесточению блок-сополимерных цепей и относительному росту их «хрупкости», что способствует разрыву ослабленных гетеросвязей (в особенности расположенных на границах гибких и жестких блоков) и соответственно частичному распаду макромолекулярных цепей и формированию более плотной упаковки как гибких блоков, так и полимера в целом.
Наблюдаемые закономерности можно также охарактеризовать как проявления тенденции к самоорганизации ММА по мере его структурирования (роста связности). Самоорганизация относится к категории самопроизвольных процессов с обратной связью, для которых характерно понижение энтропии системы, находящейся вдали от термодинамического равновесия или существующей преимущественно в волновой среде [9]. В последнем случае одним из признаков самоорганизации является преобразование параметров реакционной или молекулярной среды (например, полимерной цепи) с возникновением соответствующей обратной связи, обеспечивающей появление или рост упорядоченности структуры или движения её компонентов. При этом максимально плотная упаковка достигается не только благодаря структурно- динамическим, но и структурно-химическим переходам, т.е. участию в процессах структурирования химического механизма перемешивания полимерных цепей и(или) сегрегации блоков различной природы, а также вследствие постепенного преобразования гетероатомных группировок в составе макромолекул в более устойчивые аналоги, содержащие меньшее число гетероатомов.
Дата добавления: 2021-06-02; просмотров: 125; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!